
Table of contents

The proportion of new chemical entities with inadequate aqueous solubility has grown steadily over the past two decades. Estimates based on the FDA Biopharmaceutics Classification System consistently place between 70 and 90 percent of compounds in active development within BCS Class II or Class IV. For a Class II compound, permeability is adequate, but dissolution is rate-limiting. For Class IV, both solubility and permeability restrict absorption. In either case, the formulation must close the gap between what the molecule is thermodynamically capable of dissolving and what actually reaches the absorptive epithelium in time.
The standard formulation responses to poor solubility include amorphous solid dispersions, lipid-based systems, nanocrystals, and co-crystallization. These approaches differ substantially in mechanism, but they share a common first event. Before supersaturation builds, before micellar solubilization proceeds, before particle dissolution accelerates, the formulation surface must establish productive contact with the surrounding medium. The first wetting of a solid or droplet surface by water, enzymes, bile salts, and GI contents is often the rate-controlling step in the entire delivery sequence, and it is rarely the parameter that receives the most design attention.
The Surface as the First Decision Point
The contact between a formulation surface and an aqueous environment is governed by surface energy. A particle or droplet with low surface energy resists wetting. Water fails to spread, and the dissolution or dispersion process starts late or incompletely. This is not a purely molecular phenomenon. It depends on particle geometry, surface roughness, porosity, residual processing aids, and the solid-state history of the material during manufacturing.
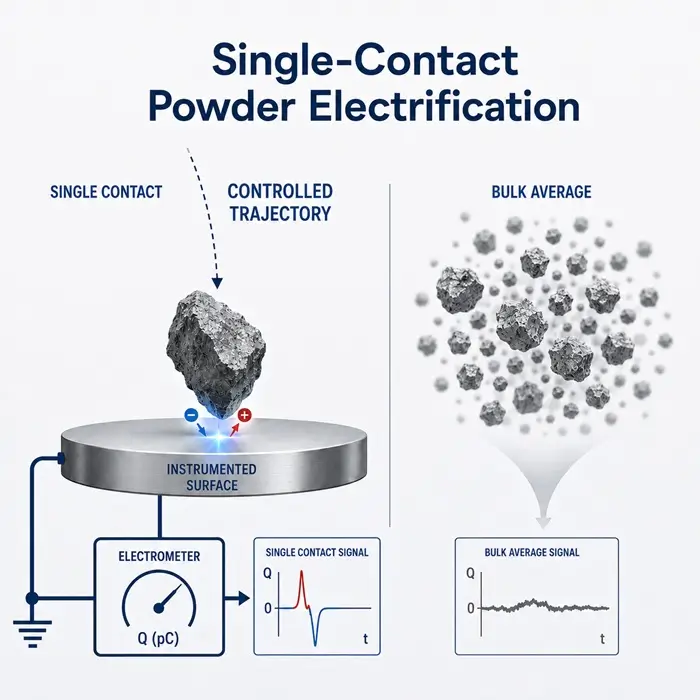
Contact angle is the most direct experimental indicator of this behavior. A contact angle above 90 degrees at the solid-liquid interface signals poor wetting and predicts slow initial dissolution regardless of the intrinsic solubility of the API. Wettability in powder systems spans a wide practical range and depends on surface chemistry, particle morphology, and the chemistry of the contacting fluid, none of which is constant across the pH gradient and enzymatic environment the formulation encounters during GI transit.
Interfacial tension between the solid or lipid phase and the aqueous medium determines how readily the system disperses. Surfactants and bile salts reduce interfacial tension and accelerate wetting, but their concentrations vary along the GI tract and between fed and fasted states. A formulation that performs well under controlled in vitro conditions can behave differently when the interfacial environment changes, and those differences are usually traceable to surface rather than bulk properties.
Molecular mobility at the surface adds another layer of variability, particularly in amorphous systems. Amorphous surfaces can undergo localized relaxation or crystallization before meaningful dissolution begins, especially after exposure to moisture or mechanical stress during processing and filling. Understanding this requires more than a dissolution profile. It requires knowing the solid-state and surface history of the API throughout the full manufacturing sequence.
Amorphous Solid Dispersions: Engineering the First Interface
An amorphous solid dispersion (ASD) suppresses the crystal lattice energy that normally limits dissolution rate. In crystalline form, API molecules are held in a periodic structure by strong intermolecular forces. Disrupting that structure, through melt extrusion, spray drying, or solvent evaporation, leaves the API in a higher-energy disordered state that dissolves faster and can achieve apparent concentrations above thermodynamic equilibrium solubility.
The surface question in ASDs is what happens when water contacts the polymer-drug matrix. The carrier must wet, hydrate, swell, and begin to dissolve in a sequence that keeps the API molecularly dispersed rather than allowing it to nucleate and recrystallize. Common polymers such as hydroxypropyl methylcellulose acetate succinate (HPMC-AS) and polyvinylpyrrolidone-vinyl acetate (PVP-VA) are selected partly for their ability to form hydrogen bonds with the API and inhibit crystallization in the supersaturated state. The relationship between polymer selection and energetic states at the particle surface determines whether that inhibition holds long enough for absorption to occur.
Research on supersaturation and liquid-liquid phase separation in ASD systems has clarified that the path from dissolution to absorption involves more than achieving high apparent solubility. The supersaturated solution can undergo liquid-liquid phase separation, generating an API-rich nanodroplet phase whose availability for absorption depends on droplet size, surface charge, and stabilization. This event can occur within minutes of dissolution onset and is rarely captured by standard sink-condition testing, which is why non-sink dissolution methods have become more relevant for ASD characterization.
The glass transition temperature (Tg) of the ASD governs physical stability. Storage above or near the Tg increases molecular mobility and raises the probability of recrystallization. Processing conditions, residual solvent content, and moisture uptake all shift the effective Tg of the dispersion. These variables interact and must be controlled throughout the full process chain, from granulation through packaging and shelf life, rather than treated as independent parameters.
Lipid-Based Drug Delivery Systems
Lipid-based formulations replace the dissolution step with solubilization. The API is either pre-dissolved in a lipid phase or intended to dissolve rapidly into it following administration. When the formulation reaches the GI lumen, it disperses into an emulsion or microemulsion under the combined action of dilution, peristalsis, bile salts, and pancreatic lipase.
The surface behavior in this context is the quality of dispersion on dilution. A self-emulsifying drug delivery system (SEDDS) generates an oil-in-water emulsion spontaneously, and the size and stability of the resulting droplets determine how much API surface area is accessible for absorption. Smaller droplets with lower polydispersity and adequate surface charge present more interface per unit volume and are less likely to coalesce or cream before absorption occurs. The HLB value of the surfactant blend and the lipid-to-surfactant ratio both determine the droplet size range achievable on dilution.
The primary risk in lipid systems is precipitation during digestion. As the formulation disperses and the lipid phase is progressively digested by pancreatic lipase, solubilizing capacity can drop faster than absorption proceeds. If the API concentration in the aqueous phase then exceeds its intrinsic solubility, nucleation and precipitation can occur rapidly and irreversibly. Polymer precipitation inhibitors, typically the same class used in ASDs, are increasingly included in lipid formulations to extend the supersaturation window through this transition.
The Lipid Formulation Classification System distinguishes simple oil vehicles from highly hydrophilic self-microemulsifying systems. As formulations move along this spectrum, the surface mechanism shifts from lipid-mediated solubilization toward mixed lipid and polymer surface behavior, with interactions between drug, excipient, bile salt, and digestive products occurring simultaneously. Selection along this spectrum should be guided by the API logP, dose, and sensitivity to aqueous precipitation, not only by ease of manufacture.
Nanocrystals: Surface Area Is Necessary, Not Sufficient
Reducing API particle size to the submicron range increases surface area per unit mass and, through the Ostwald-Freundlich relationship, raises apparent solubility relative to larger particles. The appeal is direct: more surface area means faster dissolution, and faster dissolution means more drug available for absorption within the GI transit window.
In practice, surface area is a necessary but insufficient condition. The nanocrystal surface must wet rapidly and completely, and the resulting suspension must remain stable against aggregation and Ostwald ripening long enough for absorption to occur. A poorly wetted nanocrystal surface aggregates, and the effective surface area reverts toward that of the unsized starting material. The dissolution advantage from size reduction disappears without adequate surface stabilization.
Stabilizers, typically combinations of polymers and surfactants such as poloxamers, HPMC, or PVP, adsorb onto the nanocrystal surface during wet milling or antisolvent precipitation and provide steric and electrostatic barriers against aggregation. Zeta potential gives a practical measure of electrostatic stabilization. Magnitudes below approximately 20 mV in aqueous suspension generally indicate inadequate colloidal stability, though the threshold depends on the specific system and should always be interpreted alongside particle size and polydispersity data.
Interpreting the full particle size distribution matters more here than tracking a single D50 value. The D10 and the presence of a fine tail in the distribution reflect whether the milling or precipitation process has generated the intended submicron fraction. Bimodal distributions and oversize populations indicate process inconsistency and predict performance variability batch to batch. The measurement conditions during laser diffraction require particular attention for nanosized pharmaceutical materials, where the choice of dispersion medium and stabilizer concentration can shift apparent size significantly and obscure real differences between batches.
Co-crystals and Solid-Form Engineering
A co-crystal pairs the API with a pharmaceutically acceptable coformer in a defined stoichiometric ratio, forming a new crystal structure with different intermolecular contacts than either component alone. Unlike salts, co-crystals do not require ionizable groups on the API. Unlike polymorphs, they represent a compositionally distinct solid form. The solubility advantage depends on the energetics of the new lattice: if coformer-API interactions in the crystal are weaker than the API-API interactions in the pure crystalline drug, the co-crystal will dissolve more readily.
However, this advantage is sometimes more apparent than real under biologically relevant conditions. Co-crystals can generate supersaturation transiently and then revert to the stable crystalline API form, a behavior that parallels the supersaturation dynamics seen in ASDs. The interplay of dissolution mechanisms in poorly soluble drug systems underlines that transient supersaturation only translates to improved bioavailability when the dissolved state is maintained long enough for membrane transport to occur. In vitro dissolution profiles that capture only early time points can significantly overstate the in vivo relevance.
Stability and processability constrain which co-crystal systems advance in development. Co-crystals that are metastable relative to the parent drug may convert during milling, granulation, compression, or storage at elevated humidity. A co-crystal that shows strong dissolution in simple aqueous media may behave differently across the pH and ionic strength conditions found at different GI segments. These transitions are surface events: nucleation of the stable crystalline form begins at the dissolving co-crystal surface, and its rate depends on local supersaturation, the presence of seed particles, and the surface energy of the converting form.
What Formulation Teams Should Measure
The measurement panel for a poorly soluble API formulation should be built around the decisions the team needs to make, not around instrument availability. The same measurement applied at the wrong development stage or interpreted without reference to the mechanism being tested adds data without adding confidence.
Contact angle and surface energy characterize initial wetting behavior and help explain why two formulations with similar particle size distributions perform differently in dissolution. Sessile drop measurements or the Washburn capillary penetration method can both be applied to pharmaceutical powders. The choice of probe liquids and sample preparation conditions significantly affects reproducibility and should be fixed early and applied consistently across batches and scale-up steps.
DSC identifies glass transition temperature in amorphous systems, detects residual crystallinity, and reveals polymer-drug miscibility through deviations in Tg from the calculated mixture value. XRPD provides a direct measure of crystalline content but carries detection limits typically in the 5 to 10 percent range by weight, which means meaningful crystalline content can go undetected. Both methods are necessary for ASD characterization; neither alone is sufficient. A broader framework of characterization techniques and their connection to process decisions is useful for teams building out a testing platform beyond the compendial minimum.
For nanocrystal and lipid-based emulsion systems, zeta potential provides a practical colloidal stability indicator, while particle or droplet size by dynamic light scattering tracks aggregation during storage and dissolution. Non-sink dissolution conditions are more informative than standard sink methods for ASD and co-crystal systems, because supersaturation behavior is only visible when the test is designed to replicate the concentration gradients the formulation actually encounters in vivo. Standard sink conditions mask the recrystallization events that matter most for predicting clinical performance.
DVS characterizes moisture uptake as a function of relative humidity and identifies the critical humidity threshold at which the formulation begins to absorb water rapidly. This directly predicts stability risk for ASDs, where moisture reduces Tg and accelerates physical instability, and for co-crystals, where humidity-driven phase conversion can occur at storage-relevant conditions. The relevance of moisture characterization for pharmaceutical powder formulations extends well beyond hygroscopicity classification and into processing environment control, packaging specification, and shelf-life modeling.
Manufacturing Consequences of Surface Engineering
Surface engineering for solubility enhancement consistently changes the powder processing behavior of the API in ways that deserve attention during process development. ASDs produced by spray drying or hot-melt extrusion are typically less dense, more porous, and mechanically weaker than crystalline API particles. Their flowability is often poor, and spray-dried materials can carry significant electrostatic charge from the drying process, affecting fill accuracy and creating handling challenges at commercial scale. The amorphous surface is also more susceptible to localized recrystallization during milling or blending, where mechanical activation can generate sufficient heat to shift the solid-state at the point of contact.
Flowability of ASD intermediates should be tested at the relative humidity and temperature conditions of the manufacturing environment, not only under standard laboratory conditions. Cohesive behavior increases substantially at elevated humidity, and a material that flows adequately at 20 percent RH may arch or bridge at 40 percent, a condition well within the range of commercial production facilities that do not maintain tight climate control throughout the process train.
Nanocrystal suspensions introduce different challenges. If converted to a powder for solid dosage manufacture, the nanocrystals must be redispersed without irreversible aggregation. The stabilizer system designed to protect the suspension during milling must also function through drying and redispersion. Spray drying at the nanoscale offers one route for converting nanocrystal suspensions to dry powder intermediates while preserving size distribution, though inlet temperature, feed concentration, and atomizer design all influence whether the nanostructure survives the process intact.
Lipid-based systems introduce segregation risk in blended formulations and wetting challenges at granulation or filling stages. Lipid excipients can migrate under temperature cycling during storage, altering the surface composition of the dosage unit and shifting the dissolution profile over shelf life. Monitoring surface composition, not only bulk composition, is the practical way to detect this before it reaches the patient. The broader discipline of particle engineering in pharmaceuticals has developed specific manufacturing platforms for each of these strategies, but selecting a platform without characterizing the downstream powder behavior introduces process risk that formulation testing alone cannot identify.
Solubility Enhancement Is a Surface and Process Problem
The formulation technologies that address poor solubility are scientifically mature. Their mechanisms are documented, their failure modes are known, and approved products exist for each approach. What is less reliably managed is the connection between the molecular formulation decision and the surface, particle, and process behavior that determines whether the intended performance is delivered at scale and in the clinic.
A well-designed ASD with appropriate polymer selection and a compelling in vitro dissolution profile can fail if the spray-dried intermediate is processed in a way that introduces crystallinity, if moisture control during manufacturing is inadequate, or if scale-up shifts the residence time in the extruder and changes the solid-state of the dispersion. These are surface and process failures that the formulation design assumed away, and they are often only identified after the clinical performance gap has already appeared.
The platform choice determines which surface mechanism the formulation relies on. Process control and the manufacturing environment determine whether that mechanism functions as intended at the scale and frequency required for commercial supply. Formulation scientists, analytical chemists, and powder engineers working from shared characterization data, from early candidate selection through process validation, are the combination most reliably able to close that gap before it becomes a clinical or commercial problem.






{kind=link}
{kind=link}
{kind=link}